Cancer cells are universally disturbed in their electronic energy balance, an understanding that potentially revolutionises cancer therapy and prevention Dr. Mae-Wan Ho
We are losing the war on cancer, targeting specific cancer gene mutations does not work, and for good reasons (see [1] Personalized Medicine for Cancer Fact or Fiction? SiS 54). Not only are the mutations remarkably diverse, differing between individuals and between parts within a single tumour, cancer cells soon become resistant to new drugs.
There is growing realization that cancer is not primarily a genetic disease, but an epigenetic response to chronic stress [2] (Cancer an Epigenetic Disease, SiS 54). Redundancy in diverse signalling pathways means that many different ‘adaptive’ mutations can enable cells to survive and multiply, predisposing them to malignant transformation.
One approach to cancer therapy is the much touted ‘personalized medicine’ that tailors the cure to key genes that have gone awry. But genetic heterogeneity poses a considerable, if not insurmountable hurdle [1].
The other approach is to target the most general characteristic of cancer cells and tumours that is distinct from normal cells, and this is becoming popular. Cancer cells typically have an abnormal energy metabolism, prompting some researchers to suggest that cancer is a metabolic disease [3, 4].
I prefer to call cancer a redox disease, as explained later, to distinguish it from the usual “inborn errors of metabolism” that underpinned the hypothesis of “one gene one enzyme” of biochemical genetics [5].
The abnormal energy metabolism of cancer cells was discovered by German physiologist Otto Heinrich Warburg in the 1920s. Normal cells obtain energy by breaking down the 6-carbon molecule glucose into two 3-carbon pyruvate molecules in a series of reactions – glycolysis - that does not require oxygen, followed by oxidation reactions in the mitochondria in which oxygen is needed.
Cancer cells, however, depend heavily on glycolysis to obtain energy, even though plenty of oxygen is present. This phenomenon – aerobic glycolysis subsequently known as the Warburg effect - prompted Warburg to propose that mitochondrial dysfunction was the primary cause of cancer [6].
As glycolysis is much less efficient in extracting energy from glucose, cancer cells are voracious for glucose, and that is how tumours are detected by positron emission tomography (PET) imaging in which glucose uptake is measured by means of a radioactive analogue, flourodeoxyglucose.
Aerobic glycolysis is a robust hallmark of most tumours; it involves a high uptake of glucose with lactate production in the presence of oxygen, lactate being the by-product of pyruvate, even in those cancer cells that appear to have working mitochondria [3]. The reason seems to be that cancer cells need glycolysis to generate carbon skeletons for the synthesis of proteins and nucleic acids to support rapid cell proliferation [7]; and blocking glycolysis does appear to inhibit cancer cells [8] (though it would also affect normal cells).
Warburg’s idea fell into disfavour as the view of cancer as a metabolic disease was gradually displaced with one of cancer as a genetic disease caused by mutations in specific cancer related genes, or oncogenes [3].
In recent years, the idea that cancer is a metabolic disease has become fashionable again. Some commentators remark that [4] “molecular biology is re-discovering biochemistry”; it is more important than that.
Cancer is a disease of electronic energy imbalance, and electronic energy is the life-wire that animates cells and organisms, as the father of biochemistry Albert Szent-Györgyi had discovered three quarters of a century ago [9].
In [10] The Rainbow and the Worm, The Physics of Organisms (ISIS publication) first published in 1993, I presented theoretical and empirical evidence for the quantum electrodynamic nature of organisms. An organism is energized by electrons (and protons) flowing through a liquid crystalline matrix that extends into the interior of every single cell. The movement of electrons between chemical species is reduction (for the electron acceptor) and oxidation (for the electron donor). Reduction and oxidation always go together, hence ‘redox’ reactions. Redox reactions are the heart of energy transduction in living organisms. Electrons move according to the reduction potential (also referred to as reduction-oxidation potential or redox potential), the affinity of a substance for electrons. The redox potential for each substance is compared to that of hydrogen, which is set arbitrarily to zero at standard conditions of 25 °C, 1 atmosphere, and 1 M concentration.
Substances that have positive redox potentials accept electrons from hydrogen, becoming reduced, while substances that have negative redox potentials donate electrons to hydrogen, becoming oxidized.
In order to appreciate the redox theory of cancer, we need to understand the core metabolic reactions common to organisms. For a more thorough description of energy metabolism see [11] Living Rainbow H2O (ISIS publication), a sequel to the Rainbow Worm [10] and a unique synthesis of the quantum physics and chemistry of water as the “means, medium and message” of life.
All air-breathing animals, human beings included, depend on oxygen to extract energy from their food in a universal set of core metabolic reactions (Figure 1). The 6-carbon molecule glucose is activated by ATP and the enzyme hexokinase, and split through a series of glycolytic reactions each catalysed by a specific enzyme into two 3-carbon pyruvate that take place in the cytoplasm, and do not require oxygen. Further metabolism of pyruvate normally takes place in the mitochondria, in which pyruvate is first oxidized by the enzyme complex pyruvate dehydrogenase and converted into a two-carbon fragment joined to co-enzyme A (acetyl-CoA) with the release of one CO2 and water. Acetyl-CoA enters the citric acid cycle, where it is eventually fully oxidized into further molecules of CO2 and water, generating reduced electron carriers. The reduced electron carriers shuttle electrons down the oxidative electron transport chain (ETC), and the energy released goes to make ATP (adenosine triphosphate), the universal energy intermediate in living cells.
The oxidation of glucose into carbon dioxide and water is respiration, the reverse of photosynthesis in green plants, algae and blue green bacteria. Photosynthesis captures energy from sunlight to ‘fix’ or reduce carbon dioxide from the atmosphere into carbohydrates (glucose) using electrons (and protons) obtained by splitting water, releasing oxygen back into the atmosphere in the process. The regeneration of oxygen is just as important as sequestering carbon dioxide, if not more so as far as air-breathing organisms are concerned (see [12] O2 Dropping Faster than CO2 Rising, SiS 44).
Water splitting and reforming is the redox dynamo, the magic roundabout that creates practically all life out of inanimate substances [11].
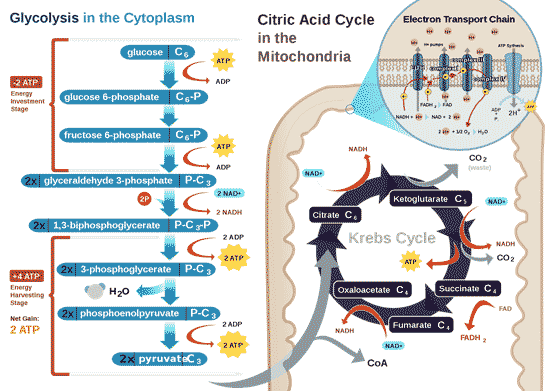
Figure 1 Energy metabolism in normal animal cells, by RegisFrey Wikimedia
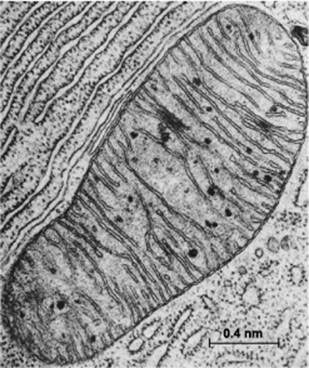
Mitochondria are special membrane-bound organelles that serve as ‘powerhouses’ in the cell (Figure 2). A mitochondrion has an outer membrane enclosing the entire structure, and a much-folded inner membrane that encloses a matrix, projecting numerous thin plate-like folds or cristae into it. Between the two membranes is a labyrinthine intermembrane space. Each mitochondrion also has 5 to 10 circular molecules of mitochondrial DNA that are replicated and inherited independently of the cell’s genome.
 .
.
Figure 2 Electron micrograph of a mitochondrion in a cell of the bat pancreas, by Keith Porter
The outer membrane of the mitochondrion contains many complexes of integral membrane proteins that form channels through the membrane, where a variety of molecules can move in and out of the mitochondrion. The inner membrane contains 5 complexes of integral membrane proteins of the oxidative electron transport chain: NADH dehydrogenase (Complex I), succinate dehydrogenase (Complex II), cytochrome c reductase (Complex III), cytochrome oxidase (Complex IV), and ATP synthase (Complex V) (see Figure 3).
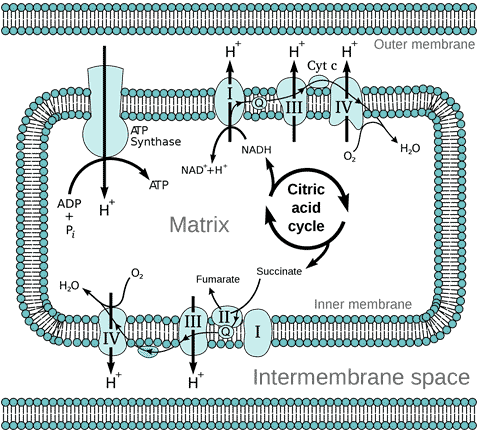
Figure 3 Diagram of the oxidative electron transport chain in the mitochondrion, by Fvasconcellos, Wikimedia
The matrix of the mitochondrion contains a mixture of enzymes that catalyse the citric acid cycle (also called the Krebs Cycle, after British biochemist Hans Krebs who discovered it). The citric acid cycle produces the electron donors NADH (reduced nicotinamide adenine dinucleotide) and FADH2 (reduced flavin adenine dinucleotide) that feed into the electron transport chain (ETC). Electron transport down the ETC is coupled with the transport of protons H+ across the inner membrane into the intermembrane space (Figure 3), resulting in a typically negative mitochondrial potential (Dym) in the matrix across the inner membrane (as protons are positively charged). The protons are returned to the matrix via ATP synthase, resulting in the synthesis of ATP from ADP (adenosine diphosphate) and Pi (inorganic phosphate). This oxidative phosphorylation is absolutely essential for the life of all air-breathing animals. Most of the ATP is produced by oxidative phosphorylation in the mitochondria. The complete oxidation of glucose generates 36 molecules of ATP, of which 32 are produced within the mitochondria, and only 4 by glycolysis in the cytoplasm.
However, glycolytic reactions are much faster. It is estimated that in the time it take for the mitochondria to produce 36 molecules from one glucose, another ten glucose molecules are turned into lactate with the generation of 20 additional ATP molecules in the cancer cell, making a total of 56 ATP molecules compared to the 36 in a normal cell [13].
Cancer cells not only exhibit aerobic glycolysis, they resistance apoptosis (cell suicide), a fate that would normally befall cells with dysfunctional mitochondria. It thus appears that aerobic glycolysis and apoptosis are linked.
Evangelos Michelakis and his team at University of Alberta in Canada were among the first to note that aerobic glycolysis and apoptosis meet up in the mitochondria [14]. They demonstrated the remarkable therapeutic potential of a cheap, readily available chemical dichloroacetate (DCA) that reactivated the gate-keeper enzyme for oxidation in the mitochondria, pyruvate dehydrogenase (see [15] Does DCA cure cancer? SiS 54), and as a result the cancer cells committed suicide and the human tumour grown in cancer-prone rats shrank. We shall look at his results in some detail, as they are relevant to our understanding of cancer as a redox disease.
The link between glycolysis and apoptosis is apparent, as many glycolytic enzymes also regulate apoptosis, while several oncoproteins induce the expression of glycolytic enzymes. This web of circular causation is what one has come to expect as a consequence of the fluid genome [16] Living with the Fluid Genome (ISIS publication), which also makes therapeutic interventions based on single molecular targets often ineffective, if not also fraught with side-effects.
The protein Akt, for example, which stimulates glycolysis and induce resistance to apoptosis, also activates hexokinase, an enzyme catalysing the first and irreversible step in glycolysis (see Fig. 1) in which glucose is phosphorylated by ATP to glucose-6-phosphate. Akt induces the translocation of hexokinase - normally residing in the cytoplasm - to the mitochondrial membrane via its downstream mediator, glycogen synthase kinase 3 (GSK3). In the mitochondrial membrane, hexokinase binds to the voltage-dependent anion channel (VDAC), an important part of the mitochondrial transit pore that controls the permeability of the mitochondria to small hydrophilic molecules. This suppresses apoptosis, presumably by making the mitochondrial membrane impermeable. Inhibiting GSK3 in cancer cells presumably causes hexokinase to unbind from the VDAC, making the mitochondria permeable to small molecules, thereby inducing apoptosis and increasing sensitivity to chemotherapy.
This suggested to Michelakis’ team that perhaps the metabolic phenotype in cancer is due to a remodelling of the mitochondria that suppresses (or disturbs) oxidative phosphorylation, enhances glycolysis and stops apoptosis.
In keeping with this hypothesis is the observation that cancer cell lines have more hyperpolarized mitochondria membrane potential (more negative compared to the outside) (see Box 1) [17]. Cancer cells also relatively deficient in the cell membrane voltage-gated K+ (Kv) channels (channels for K+ that open only if the electrical potential is beyond a threshold value). K+ channel deficiency is known to suppress apoptosis in several cell types including cancer cells.
Box 1
Hyperpolarized (more negative than normal) mitochondrial electric potential Dym has been linked to malignant transformations since the 1980s. Tumours cells are typically highly heterogeneous, and within a population of tumour cells, there are minor subpopulations with stable differences in their Dym that survive cell cloning. Cells with high Dym typically have decreased sensitivity to chemoprotective agents and increased secretion of VEGF (vascular endothelial growth factor, promoting growth of blood vessels), and in metastatic tumours, but not in non-metatstatic tumours, correlated with invasive potential [17].
However, mechanisms involved in generating and maintaining difference in Dym are unclear, they may reflect alterations in the composition of the mitochondrial membranes, modulations in expression of mitochondrial targeted nuclear genes, or enrichment in a particular mitochondrial population.
Treatment with DCA decreased the hyperpolarized mitochondrial potential to normal levels, accompanied by a decrease in tumour cell growth in vitro and in vivo, as reported [15].
The mitochondrial potentials in three human cancer cell lines: A549 (non-small-cell lung cancer), M059K (glioblastoma), and MCF-7 (breast cancer), were compared with healthy, noncancerous human cell lines: small airway epithelial cells (SAEC), fibroblasts and pulmonary artery smooth muscle cells (PASMC). All cancer cell lines had significantly more hyperpolarized mitochondrial potential compared to normal cells, as measured by increased fluorescent of the potential sensitive dye tetramethyl rhodamine methyl ester TMRM. Incubation of all three types of cancer cells with DCA reversed the hyperpolarization and returned it to the level of normal cells after 48 h. But normal cells were unaffected. The DCA effects on mitochondrial electric potential occurred as quickly as 5-10 min and were dose dependent.
The DCA-induced decrease in electrical potential of the mitochondria was limited by an inhibitor of the VDAC; indicating that transport out of the mitochondria is important for the DCA response. As consistent with this hypothesis, DCA caused the efflux of pro-apoptotic factors from the mitochondria, as well as increased reactive oxygen species production (see below). In untreated A549 cells, cytochrome c and the proapoptosis inducing factor (AIF) were restricted to the mitochondria. But in DCA treated cells, cytochrome c was diffusely present in the cytoplasm and AIF was translocated to the nucleus, both indicative of apoptosis.
Moreover, DCA increased glucose oxidation by 23 % and concomitantly suppressed glycolysis and fatty acid oxidation in A549 cells. After 48 h of DCA treatment, the extracellular lactate level was decreased, while pH increased in A549 cells compared with untreated cells.
Reactive oxygen species (ROS) are small molecules containing oxygen that are more reactive than ordinary molecular oxygen. ROS are produced in mitochondria as intermediates of electron transport [18] (see Box 2).
Box 2
In the process of oxidative phosphorylation, oxygen is reduced one electron at a time in a sequence, oxygen to superoxide to hydrogen peroxide to hydroxyl radical, and finally water:
O2 → O2-· → H2O2 → ·OH → H2O
All except the first and last have an unpaired electron, and are very reactive, hence referred to as reactive oxygen species (ROS). Thus, oxidative phosphorylation inevitably generates ROS as intermediates, and the mitochondria are considered the major source of ROS; the primary ROS being superoxide anion, O2-·. It is the precursor of all ROS species, and in vivo it is produced both enzymatically by NADPH oxidase, and xanthine oxidase, and non-enzymatically, when a single electron is directly transferred to O2. The superoxide anion acquires a proton to become a hydroperoxyl radical (H O2-·), followed by a fast rearrangement (dismutation) either spontaneously or through a reaction catalysed by superoxide dismutases (SODs) to produce hydrogen peroxide H2O2. H2O2 is relatively stable and membrane permeable; and can diffuse within the cell to be eliminated by antioxidant systems in the cell or mitochondria, such as catalase, glutathione peroxidase, and thioredoxin peroxidase.
There is disagreement as to whether normally functional mitochondria actually export ROS [18, 19]. I believe it is entirely possible that ROS is only produced as the result of diminished coherence in electron transport, resulting in partially oxidized intermediates, because that’s what ROS consist of. DCA increased the production of the ROS hydrogen peroxide (H2O2) in a dose-dependent manner from 25 % at 0.05 mM DCA to 35 % at 0.5 mM DCA [14]. This increase was inhibited by rotenone, suggesting the involvement of complex I of the electron transport chain, presumably in a reversed electron transport due to a build-up of NADH, another sign that the mitochondrial ETC is not functioning normally in cancer cells. Consistent with this hypothesis is the observation that isolated mitochondria exposed to DCA showed an increase in NADH levels within the mitochondria [19].
One complication is that ROS at lower levels, characteristic of chronic stress and inflammation, are a ‘second messenger’ for cell proliferation - a predisposition to malignant transformation - supporting the idea that cancer is an epigenetic disease [2]. However, the evidence linking mitochondrial ROS, presumably at higher concentrations, to apoptosis is equally strong.
The main ROS produced in mitochondria is H2O2 (see Box 2). If it is not eliminated by the cell’s antioxidant system, it can be further transformed to hydroxyl radical (·OH) in the presence of metal ions. ·OH is highly reactive, and damaging [18]. A wide range of mitochondrial ROS-induced damages has been described, to proteins, lipids and mitochondrial DNA. These damages can result in an energetic catastrophe.
As described by Michelakis’ team [14], the major ROS target inside the mitochondria is the permeability transition pore, which becomes highly conductive in the presence of ROS, allowing small molecules to pass in both directions. Small solutes flood into the mitochondrial matrix along their electrochemical gradients (from high concentrations outside to low concentrations), dissipating the electrochemical potential and inducing swelling of the mitochondrial matrix, eventually rupturing the outer membrane, releasing cytochrome c and proapoptosis inducing factor (AIF) into the cytoplasm, resulting in apoptosis. Cells use a special form of autophagy, mitophagy [18], to selectively eliminate defective mitochondria. Increases in cellullar ROS leads to loss of mitochondrial membrane potential, which is a trigger for mitophagy. When many mitochondrial are eliminated by mitophagy, apoptosis follows.
Increase in H2O2 production by cancer cells exposed to DCA is involved in activating voltage gated K+ channels (Kv) in the cell membrane. Michelakis team showed that DCA treatment increased the K+ outward current significantly in all cancer cells but not in normal cells [15]. This increase in outward K+ current, accompanied by an increased expression of the K+ channel Kv1.5, leads to hyperpolarization of the plasma membrane (becoming more negative); and is blocked by intracellular catalase, which breaks down H2O2, and by rotenone which inhibits complex I produced H2O2.
At the same time, DCA decreased intracellular Ca2+ by inhibiting voltage-gated Ca2+ channels, so DCA treated cells had lower intracellular Ca2+ compared with untreated cells, the decrease occurring within 5 mins and was sustained after 48 hours of DCA exposure. The effects on Ca2+ were inhibited by rotenone and mimicked by H2O2, among other things.
DCA is thought to decrease intracellular Ca2+ and increase Kv1.5 expression by inhibiting NFAT (nuclear factor of activated T lymphocytes). NFAT is known to inhibit both apoptosis and the expression of Kv1.5 in myocardial cells, and the team found that this was also true in cancer cells. Increase in intracellular Ca2+ activates calcinerin, which dephosphorylates NFAT, allowing it to be translocated to the nucleus where it regulates gene transcription. DCA-induced activation of Kv1.5 leads to hyperpolarization of the cell membrane, inhibiting voltage gated Ca2+ channels, hence blocking the increase in intracellular Ca2+ and inhibiting NFAT.
Michelakis and colleagues found that DCA increases annexin expression, caused a ~6-fold increase in TUNEL-positive nuclei and activates both caspase 3 and 9 in A549 cells. Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) is a method for detecting DNA fragmentation by labelling the terminal end of nucleic acids.
DCA appears to eliminate highly proliferative cells by inducing apoptosis and by decreasing intracellular Ca2+ levels. It also decreases cell proliferation, as measured by BrdU (bromodeoxyuridine) incorporation, and the expression of proliferating cell nuclear antigen (PCNA). In addition, DCA decreased the expression of survivin, a mitotic indicator.
DCA induces apoptosis of cancer cells by two pathways, one in the mitochondria, where depolarization activates mitochondria-dependent apoptosis, and the other at the cell membrane, where upregulation of Kv1.5 channels decreases K+, activating caspases. The mitochondrial component is thought to be more important, as other factors and manipulations to deliver the cytoplasmic component of apoptosis did not result in the degree of apoptosis induced by DCA.
These findings, in addition to the demonstration of the ability of DCA to shrink xenograft human tumours in rats, and glioblastomas in humans [16] do support Warburg’s hypothesis that cancer is a disease involving mitochondrial malfunction; but perhaps not in its original form, as Warburg thought mitochondrial were totally inactive.
Not much attention has been paid to the electronic state of the cell or its organelles until quite recently when voltage sensitive dyes became available. This made it much easier to measure the electric potential of cells and organelles. As a result, researchers discovered that the cell’s electric potential determines its vital states, from cell division and pattern formation to differentiation, regeneration and cancer ([20] Membrane Potential Rules, SiS 52). This is fully in accord with the quantum electro-dynamic nature of life [10, 11].
Actually, it has been known since the 1950s that the cell membrane potential, measured with microelectrodes, varies throughout the cell cycle [21]. Cell types with very high resting potentials such as muscle cells and neurons show little if any tendency to divide, while a decrease in membrane potential follows malignant transformation. In the 1970s, Clarence D. Cone Jr. induced DNA synthesis and mitosis in fully differentiated neurons from the central nervous system using a variety of agents that depolarized the cell membrane (made it less negative) [22]. In the 1990s, electric potential measurements of skin sites over malignant tumours of the breast gave electropositive readings that were correlated with increased depolarization in membrane potential of cancerous cells and tissues compared with normal cells or non-cancerous cells [23].
The other well-known sign of redox imbalance in cancer cells is the hyperpolarized mitochondria (see Box 1).
Additional evidence is now coming from direct measurements of redox states. The redox pairs of the cell are NADH/NAD+ (nicotinamide adenine dinucleotide), NADPH/NADP+ (nicotinamide adenine dinucleotide phosphate) and GSH/GSSG (glutathione). The ratio of reduced to oxidized forms reflect the redox state of the cell. For example, under unstressed conditions in cultured astrocytes (brain cells that control blood flow to neurons), the NADH/NAD+ pair is predominantly in the oxidized state to accept electrons produced during glycolysis in the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) reaction (see Fig. 1). In contrast, the redox pair NADPH/NADP+ are kept in a more reduced state to provide electrons for reductive biosynthesis, while the concentration of GSH strongly exceeds that of GSSG to support efficient antioxidant defence. These ratios of the redox pairs in cultured astrocytes are similar to those reported for brain [24], and are intimately linked to cellular metabolism and function.
Thioredoxin - a class of small redox proteins present in all organisms that act as antioxidants with redox signalling functions - is believed to integrate the overall redox state of the cell, and are essential for life in mammals [25]. Researchers at University of Wisconsin School of Medicine and Public Health, Madison, in the United States, examined protein levels and redox changes of thioredoxin 1 (Trx1) in human prostate tissues and culture cells [26]. They found more than 4-fold increase in Trx1 protein in the nucleus of high-grade cancer cells compared with normal controls, and the increase correlated with cancer progression. The protein was also increased in the cytoplasm by about 2-fold. Despite increased protein levels, the oxidized forms of nuclear Trx1 were higher in prostate cancer cell lines compared to their benign counterparts, suggesting that nuclear redox imbalance occurred selectively in cancer cells.
Trx1 has a specific role in the modulation of redox signaling, with distinct nuclear and cytoplasmic pools, each performing different functions. In the nucleus, Trx1 interacts with certain transcription factors to regulate their binding to DNA; these include p53 (apoptosis response), nuclear factor κB (NF-κB, involved in inflammatory response) and nuclear factor-like 2 (Nrf2, involved in antioxidant response). In the cytoplasm, Trx1 can regulate apoptotic signal-regulating kinases. Trx1 is also known to move from the cytoplasm to the nucleus in response to oxidative stress. Selective oxidation of Trx1 can occur and has been detected in both the nucleus and the cytoplasm in response to cellular redox changes. Increased Trx1 protein expression has been detected in multiple cancer tissues and cancer cell lines, and an increase in Trx1 expression was associated with higher tumor grade and has been implicated in the resistance of tumor cells to certain chemotherapies and ROS generating agents.
Emerging evidence suggests that cancer cells are more oxidized relative to normal; they do not have enough electrons. This is consistent with other indications that cancer is a redox disease, a state of electronic imbalance. Rational therapy and prevention should start from here.
Article first published 12/04/12
Comments are now closed for this article
There are 19 comments on this article.
Dr. Michael Godfrey Comment left 13th April 2012 04:04:35
The whole approach to cancer prevention and treatment warrants re-evaluation in view of this fundamentally important evidence. It seems to me that recharging by regularly earthing barefoot on the ground for an hour a day might also be beneficial as this has been shown to reduce inflammation and improve capillary blood flow. (www.earthing.com).
Konstantin Comment left 13th April 2012 04:04:40
Awesome article. I´ve got one question though regarding K-Channels etc. I haven´t received Gilbert Ling´s book yet, but it was my understanding that channels don´t exist and acutally represent protein-interactions which correlates to the fluid crystal structure of the cell. Is that right?
Mae-Wan Ho Comment left 13th April 2012 04:04:36
Michael Godfrey, you are right. I was sceptical about that for a long time, but earthing does make sense. Konstantin, you raise a very good point. I deal with the issue in more detail in my new book, Living Rainbow H2O. Gilbert will have to speak for himself. But he may be right in that what conventional cell biologists regard as a potassium channel could well be something else. Also, Gilbert doesn't actually deny the existence of potassium channel as such in some specialized epithelial cells.
Anita Allen Comment left 13th April 2012 16:04:03
Your attention is drawn to Eleni Papadopulos-Eleopulos’ Oxidative Stress Theory, first proposed in 1982 as the cause of cancer (Papadopulos-Eleopulos, E. (1982) ‘A Mitotic Theory’. Journal of Theoretical Biology 96, pg 741-758) and then applied to AIDS in the a 1988 paper (Papadopulos-Eleopulos, E. (1988) ‘Reappraisal of AIDS: Is the oxidation caused by the risk factors the primary cause?’ Medical Hypotheses 25;151-162) and elaborated in another paper in 1992 (Papadopulos-Eleopulos E., Turner V.F., Papadimitriou J.M. (1992) ‘Oxidative Stress, HIV and AIDS’ Research in Immunology 143: 145-148). According to this proposal, normal cellular functioning is determined by the level and oscillations of cellular redox - oxidation and its chemical opposite reduction. When oxidation is prolonged or excessive, cells become abnormal, injured and susceptible to diseases. As far as I know, Papadopulos-Eleopolus is the first to advance the theory that cellular processes have a cyclic nature controlled by a periodic charge exchange between actin and myosin, regulated by the oxidation and reduction of sulphydryl moieties. Also, she is the first to apply oxidative stress as the mechanism leading to AIDS.
Regarding treatment, she proposes that oxidative stress requires that the patient be treated with antioxidants in general and –SH (sulphydryl) containing compounds in particular. The oxidative theory demands that the underlying cause, cellular oxidation, must be addressed – that is treatment with –SH containing compounds under the supervision of a doctor who has access to laboratory facilities to measure redox and at least until the redox is normalised. The treatment may include adjunct measures such as diet and stress reduction management. At the same time, exposure to oxidising substances should be minimised, or if possible, eliminated.
Konstantin Comment left 15th April 2012 14:02:04
Most of the stuff discussed here is still way beyond me. Science is complicated and you have to learn a lot of names :)
But one thing i start to understand is, that life is a flow of electrons and its this flow that connects all living matter. If people would start to understand this, the way we live would change tremendously. I think its really important to spread this idea, if one wants to make a positive change in this world. Basically this is how indigenous people used to understand their presence.
What do you think about violet rays, rife-machines etc.? They were eventually banned by the FDA, but there is a lot of evidence that these high-frequency gadets helped with many ailments. If electronical flow is the basic of life, "charging" the cell with these machines sounds like a good idea to me.
Ren Comment left 3rd May 2012 23:11:11
On DCA and cancer cells metabolism:
http://www.ncbi.nlm.nih.gov/pubmed/21557214
http://www.thedcasite.com/the_dca_papers.html#Conti
Danilo Comment left 1st September 2012 05:05:21
From “Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer” ( http://www.amazon.com/Cancer-Metabolic-Disease-Management-Prevention/dp/0470584920 ) by
Thomas Seyfried (Professor of biology, Boston College): “The gene theory has deceived us into thinking that cancer is more than a single disease (…) Cancer is a singular disease”.
Irucka Embry Comment left 29th September 2012 16:04:05
Is cancer caused by a fungus? See http://www.cancerfungus.com/ . Thank-you.
David Lowenfels Comment left 8th December 2012 07:07:10
May I refer you to the work of Dr.med. Heinrich Kremer who has written much about the topic of redox, mitochondria, cancer, and HIV. http://www.amazon.com/Silent-Revolution-Cancer-AIDS-Medicine/dp/1436350832
http://www.thenhf.com/old/articles/articles_554/articles_554.htm
Zoco Comment left 11th January 2013 22:10:51
Hi, Great article ! !
I have come across a so called redox therapy peoduct Asea,
Its Terribly pricy, first product of marketing company. Says biggest breakthrough ever. I think its a scam. Have you heard of it?
Budwig diet is supposed to renew redox state if cell , its lipid membranes.
Dca is great but some cancers have dual metabolism (also fatty acid) like colorectal, most leukemias and Dca makes there thing worse, forcing fatty acid oxidation instead of glycolysis.
C.DELARGE Comment left 10th May 2013 20:08:06
You should try a perfectly balanced REDOX signaling molecules stabized solution called ASEA, it is not a scam and really works,is highly patented and stabilized after many years of research.
You can have more informations or even order it for research on http://cedel.myasealive.com
I would behappy to send you all the tests conducted so far or even put you in contact with Dr Gary L. Samuelson who helped stabilize those redox molecules.
Ramachandran Comment left 24th October 2013 06:06:45
Cancer is curable! I am sure on this. The doctors should join hands with pharmacologists and herbal analysers. The persons should not try to earn a name and fame. It will come automatically provided if your mind is free from all requirements, such as money, properties etc. You should make others to understand your views and presentation. You should not argue that what you present/ invent is your own research, may be your application but a root will be somewhere else. A paper can not be presented in 20 minutes and cross examination/ verification time is another 10 mts.which is not enough for a discussion.
According to Veda woman is a magnetic force, and pulls (Man) electrons in her.
A woman has more minerals in her, that forms a baby.
Secondly you must know that the uterus of woman plays major role in forming and curing cancers. For male it is mind and body. For any diseases in woman try to study the uterus first. The medicine you give to a woman first goes to liver forms amino acids and then to uterus, supported by Ascorbic acids and mix with blood.
For male you should concentrate on belle button and diet.
One thing is common here. A medicine you give in South America will not work with Indian climate and on Indians.
Finally regarding Oxygen in human, Oxygen is an emulsifier, that mixes all the natural minerals (in Vegetables, diet and atmosphere) and carries them in to the body through lungs and liver, and distribute the same in to the body.
For some patients the administrating oxygen, will dilute the minerals and there by takes long time to react, and patient will sweat a lot, or/ and pass more urine, and this may give a load of work to the kidneys.
If you start another research based on the above mentioned information I am ready to share my findings with you.
--
Regards
Ramachandran
Bob Wheeler Comment left 11th May 2014 14:02:59
ASEA is enhancing my vitamin C IV therapy as I fight stage 4 rectal cancer and prostate cancer. Also a third primary cancer in my colon. I am doing many things, Gerson diet as a base and 12 to 16 ounces of ASEA daily. We are making headway. I am looking at magnetic pulse and electro/light therapy very seriously. This is a great article!
Sarah Wright Comment left 25th May 2014 03:03:50
Interesting article. I'm sure this is the right path to our understanding
However we must also look at what is causing this redox imbalance
Anyone with cancer must detox. That also means removing any wireless transmitters such as cordless landlines and Wifi otherwise the cancer will just come back.
www.bioinitiative.org
Noe Comment left 23rd August 2014 07:07:04
Have you or any one have make a research about the product called ASEA?? and how they claim that it has redox? Wouldn't that be a good thing?
Lorraine Wright Comment left 23rd March 2015 19:07:08
Should be possible to kill cancer cells and reduce tumours by using homeopathic remedies of dichloroacetate (MM potency) together with hydrogen peroxide (MM potency) and thioredoxin (MM potency). Homeopathy can be used to correct excesses as well as deficiencies in the physical body. This will also help correct other disease states where inflammation is an issue and redox imbalances ensue. I am currently using carbon monoxide, nitric oxide and hydrogen sulphide in potency to correct redox signaling problems with immediate, positive results in energy levels and thought processes.
Lynn Matthews Comment left 26th September 2016 20:08:57
Excellent article. The scientific and medical communities are coming more into agreement that oxidative stress and redox imbalance are potentially the root cause of a high percentage of chronic diseases present today. ASEA redox supplement is currently the only technology available that delivers balanced, stabilized bio-active redox molecules. Clinical results demonstrated it to significantly reduce oxidative stress in participants in just 12 weeks.
Ronnie Cumbie Comment left 22nd September 2016 23:11:15
Great article! Stabilizing redox signaling molecules outside the body is exciting as well. And apparently, the only product that does this is ASEA I discovered it about six months ago, and I think we may have found the true 'Fountain of Youth.'
Dr.Soumitra Kumar Choudhuri Comment left 22nd September 2016 23:11:53
Article is relevant, lucid and well illustrated.