New Genetics & Evolution
Non-coding DNA in genomes increases in concert with the increase in developmental complexity in evolution, and is consonant with the important regulatory roles identified for the many classes of non-coding RNAs transcribed from more than 85 % of the DNA regarded as ‘junk’ not so long ago Dr. Mae-Wan Ho
It wasn’t so long ago that most people still believed DNA carries the instructions for making an organism, while RNA simply copies out (transcribes) the instructions (by complementary base pairing) that are then translated into protein via a genetic code, in which different triplets of bases (codons) specify one of twenty amino acids plus start and stop signals. The proteins are the real workhorses in this hierarchy, with the DNA akin to the Holy Scripture – ‘Book of Life’ the Central Dogma – faithfully copied and transmitted by scribes (RNA), to be interpreted and implemented by the faithful (proteins).
But soon after the human genome sequence was announced, it became clear that RNA plays a much more substantive, central role than previously thought.
In [1] (Subverting the Genetic Text, SiS 24) published in 2004, I wrote of “the hidden intrigues in the vast RNA underworld where layer of interference and machinations subvert the chain of command from DNA to RNA to protein”, and “RNA agents not only decide which bits of text to copy, which copies get destroyed, which bits to delete and splice together, but also which copies are to be transformed into a totally different message, and finally which resulting message – that may bear little resemblance to the original text - gets translated into protein.” And most outrageously, “RNAs even get to decide which parts of the sacred text to rewrite or corrupt.” This picture is now being fleshed out in minute detail.
RNA plays a central role in the natural genetic modification at the heart of living processes that change the genetic information of cells and organisms as they experience their environments that are necessary for survival, and some of which are passed on to the next generation(s) [2]. By genetic information, I include changes in genomic DNA sequence -conventionally designated ‘genetic’ - as well as changes in RNA and chemical markings of DNA, histones, and RNA - generally designated ‘epigenetic’. Not only do they form a seamless continuum, but RNA is also independently replicated and inherited, as is now known. ‘Natural genetic modification’ is hence broader concept than James Shapiro’s ‘natural genetic engineering’, which is restricted to changes in genomic DNA sequence [3-5] (see [6] Evolution by Natural Genetic Engineering, SiS 63).
The RNA underworld is truly vast, and it is taking an army of researchers using the most sophisticated next generation ‘deep sequencing’ technologies to decode (see Box in [7] Nucleic Acid Invaders from Food Confirmed, SiS 63). New species of RNA, with highly specific and esoteric functions are being discovered every day.
Proteins are a tiny minority when it comes to executing biological functions. To get a true sense of proportion, the human genome contains only about 20 000 protein-coding genes, similar to nematodes that have only 1 000 cells compared to humans’ 1014 cells. In contrast, non-protein-coding DNA increases with increasing complexity, reaching 98.8 % of the human genome, much of which referred to as ‘junk DNA’ until geneticists discovered to their surprise that most of these sequences, (> 85 % according to latest estimate [8]) are dynamically transcribed, mainly into non protein-coding (nc)RNAs, apart from ribosomal RNA and transfer RNAs (rRNAs and tRNAs) that play crucial roles in translation of the mRNA into protein.
John Mattick, director of the Garvan Institute of Medical Research in Sydney, Australia, is especially impressed with the tens if not hundreds of thousands of long ncRNAs (lncRNAs) showing specific expression patterns and subcellular locations, plus many classes of small regulatory RNAs (sRNAs) that have already been discovered. In a comprehensive review published in 2011, he argued that the increase in genome size in evolution is causally linked to the increase in developmental complexity of organisms [9]. Specifically, the extra ncDNA is transcribed into the multitudes of ncRNAs that create the additional layers of developmental complexity required for the evolution of eukaryotes, multicellular plants and animals, and in particular of primates and the human brain.
Figure 1 (top) plots the proportion of ncDNA/total DNA for the major groups of organisms in order of their time of appearance in evolution. It increases in the course of evolution in jumps correlated with the major transitions in developmental complexity of organisms: from prokaryote to eukaryotes, from single celled protists to multicellular, the appearance of plants, chordates and vertebrates in succession. The bottom graph is a theoretical plot of ‘complexity’ in parallel to ncDNA/tDNA , showing more explicitly the step increases for the major evolutionary transitions, together with the key events accompanying the increases. The single cell eukaryotes evolved by acquiring wholesale symbiotic bacteria (and their genomes) to form organelles such as the chloroplasts, mitochondria and the nucleus, accompanied by a spatial separation of transcription in the nucleus from translation in the cytoplasm. The acquisition of introns (intervening non-coding sequences of split genes) and their subsequent expansion, and the evolution of splicing mechanisms coincide with the origin of the multicellular eukaryotes. A steep rise in ncRNA thereafter is associated with the elaboration of RNA-regulatory networks with all the specific signaling, interpretation and execution mechanisms.
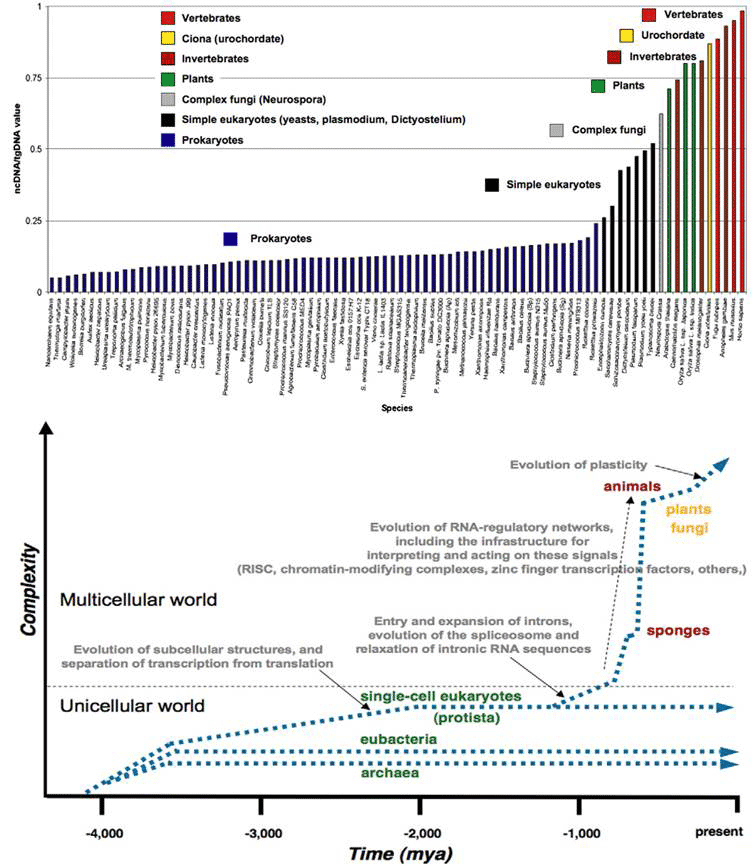
Figure 1 The increase in non-coding DNA in evolution correlates with increase in developmental complexity (rearranged from [9]
In fact, the pioneers of molecular genetics recognized that genes coding for proteins was by no means the whole story. Barbara McClintock (1902-1992), awarded the Nobel Prize late in her career, was ignored and even ridiculed for her identification of transposons (jumping genes) as ‘controlling elements’ in maize. In the late 1960s, Roy Britten (1919-2012) and Eric Davidson (now at Caltech in the United States) noted that the extent of genomic DNA broadly increased with developmental complexity and found unexpectedly that the population of ‘heterogeneous nuclear RNA’ (hnRNA) is far more complex than messenger RNA (mRNA, which code for proteins), and a substantial proportion of the genome is comprised of low complexity high copy number repeat sequences, some of which at least are specifically expressed at different developmental stages. This led them to propose that there may be considerable regulatory RNA in the nucleus of eukaryotic cells and that repetitive sequences (later found to be derived from transposons) may be part of the regulatory networks. They also predicted that many of the proposed nuclear regulatory RNAs would be associated with chromatin. They were right.
The surprises started coming in the late 1970s. The interrupted gene was discovered in 1977. Most protein coding genes in mammals and other complex organisms are mosaics of small segments of protein-coding sequences, exons, interspersed with often vast tracts of non-protein-coding sequences, introns. Introns were thought to be evolutionary relic or debris, i.e., junk DNA. A far more interesting interpretation is that the excised RNAs are also transmitting information as part of a vast RNA regulatory network.
The second surprise was that the genomes of humans and other complex eukaryotes are full of transposon-derived sequences of various classes, pejoratively referred to as ‘repetitive elements’ and assumed to be non-functional ‘selfish DNA’, part of the evolutionary junk yard. It has been estimated by neo-Darwinists that only ~5% of the human genome is under ‘purifying selection’, and anyone disagreeing with that is treated with sarcasm and hostility.
On the contrary, Mattick pointed out that the raw material for evolution is duplication of sequences and transposition to spread functional cassettes around the genome and re-structure regulatory networks accompanying the divergence of phenotype.
The protein toolkit of all organisms have remained roughly the same in billions of years, consequently, it is the regulatory architecture that makes the difference. Prokaryotic genomes are predominantly made up of protein-coding sequences, and there is a clear limit on what you can do with different combinations of protein-coding sequences. More complex eukaryotes must have solved this problem, Mattick proposed, by moving to a more genomically efficient (and evolutionary flexible) RNA-based regulatory system.
While protein-coding genes and protein-coding sequences remains surprisingly static, the extent of non-protein-coding intron and intergenic sequences does scale with increased developmental complexity (see Fig. 1), and is the only variable so far demonstrated to do so, along with the complement of regulatory RNA.
The fourth great surprise of genomic analyses is that the vast majority of non-protein coding sequences are transcribed and apparently in a developmentally regulated manner. Some ncRNAs are easily detectable by the relatively insensitive method of in situ hybridization, and found to be located in particular subregions of the brain, such as the dentate gyrus of the hippocampus thought to be involved in the formation of new episodic memory, and would not be detectable in a whole brain transcriptomic analysis.
Recent discoveries in ncRNAs fully corroborate Mattick’s thesis.
There are tens if not hundreds of thousands of long non-coding intergenic and intronic RNAs (lncRNAs , >200 nt) expressed from mammalian genomes, with abundant evidence of their involvement in cell and developmental biology. The central nervous system is by far the most complex and diversified organ in terms of ncRNAs. A large fraction of lncRNAs is expressed in the brain. Many are derived from enhancers, non-coding regulatory elements acting at considerable distances from the genes they control in expression during development. Some lncRNAs act like enhancers, suggesting that enhancer action may involve a derived RNA. There is also evidence from studies on synonymous codon sites that even mRNAs may have embedded regulatory functions in addition to their protein-coding capacity. Even conventional protein coding loci may produce both small and large ncRNAs by regulated post-transcriptional cleavage of mRNAs, including within the 3’ untranslated regions, which can be expressed in a highly cell-specific manner, as in the cortex and hippocampus of the brain, or Sertoli cells in the testis. That is why synonymous mutations (those that do not change the amino acid encoded) can make a big difference, contrary what has been generally assumed previously (see [10] New Hazards in GMOs from Synonymous Mutations, SiS 61), with large implications on the safety of GM food.
Transcriptome analysis indicated that many lncRNAs expressed in the brain are specific to primates or humans [11]. One lncRNA that has rapidly evolved since humans diverged from the other great apes is HARIA (highly accelerated region 1A). Its expression level correlates with that of reelin, a protein crucial for brain development. In contrast, other classes of brain-expressed lncRNAs seem to be highly conserved from birds to mammals and have a similar spatiotemporal expression profiles (reviewed in [12]). Moreover they are transcribed from complex genetic loci where they often overlap or are antisense to genes encoding key developmental regulator proteins. Such lncRNAs can modulate the activity of their nearby genes by acting as molecular scaffolds to recruit specific factors.
A new class of circular RNAs (circRNAs) has been identified [13] that function as sponges for microRNAs (miRNAs), tiny regulators of gene expression. One 1 500 nt species expressed in the brains of mice and humans contains about 70 binding sites for a miRNA called miR-7, targets of which have been linked to cancer and Parkinson’s disease.
lncRNAs are also known to function in the development of diverse organs and tissue types.
One of the first examples of functional lncRNA is the identification of X-inactive specific transcript (Xist) as a regulator of X chromosome inactivation in mammals, which is directly involved in the formation of repressive chromatin [14]. Xist deletion in mice causes a loss of X chromosome inactivation and female-specific lethality. XIst acts in cis - on the X chromosome from which it is transcribed - to induce the formation of transcriptionally inactive heterochromatin. Its expression is controlled by other lncRNAS in both positive and negative manner.
Similar lncRNAs are involved in imprinting. Imprinted genes generally occur in clusters and are epigenetically marked in a sex-dependent way during male and female gamete formation. They are subsequently silenced on only one parent chromosome in the embryo. Imprinted regions encode different species of lncRNAs that in many cases bind to imprinted regions and are directly involved in silencing. These lncRNAs are generally long (more than 100 kb) and function in cis.
HOX genes are an evolutionary conserved family of transcription factors that regulate the body plan during embryonic development and contribute to cell specification in adult differentiation. In mammals the 39 HOX genes are grouped in four clusters. In addition to protein-coding genes, the clusters produce hundreds of lncRNAs that show similar spatiotemporal expression to their neighbouring protein-coding genes. Some of these lncRNAs have been shown to be directly involved in regulating the HOX genes.
Several lncRNAs show expression profiles correlating with core components of the transcriptional network controlling pluripotency, and the promoters of these lncRNA species are bound by at least one of the core pluripotency transcription factors.
The vast majority of lncRNAs have yet to be characterized, but compelling genome-wide characteristics indicate that they are functional [9]. Their control elements such as the promoter, splice junctions, exons, as well as predicted structures, genome position and expression patterns are conserved in evolution. They are dynamically expressed and alternatively spliced during cell differentiation, and splicing patterns are altered in cancer and other diseases. LncRNAs are associated with specific chromatin signatures of actively transcribed genes. They are implicated in the regulation of key morphogens for pattern formation, transcription factors and hormones. And they show tissue and cell specific expression, to a much finer degree than proteins.
A survey of the expression of some 1 300 lncRNAs in mouse brain showed that over 600 were expressed in highly specific locations such as different regions of the hippocampus, different layers of the cortex, or different parts of the cerebellum with most of them showing specific subcellular locations including novel subnuclear domains in some neurons or Purkinje cells. For example, lncRNAs are found in ‘paraspeckles’ that retains RNA-edited transcripts containing Alu elements, and are induced in differentiated cells. (Alu elements are the most abundant transposable elements in the human genome, and were acquired by the genome of an ancestor of Supraprimates [15]). Preliminary evidence suggests that paraspeckles may be involved in regulating nuclear-cytoplasmic shuttling of RNAs subject to RNA editing associated with cognition, a feature not only of brain function, but also of mammalian reproduction, development and physiology associated with extended postnatal nurturing. Most functionally characterized lncRNA appear to play a role in differentiation and development, which is consistent with the progressive expansion of these transcripts in developmentally complex organisms, and with their major function in the regulation of epigenetic processes central to differentiation and development.
Epigenetic information encoded by the methylation and hydroxymethylation (gene repression and activation respectively) of cytosine in DNA, and a wide range of modifications of the histones that package DNA into nucleosomes are now well-known (reviewed in [9]). These are catalyzed by a suite of about 60 generic enzymes/chromatin modifying complexes that put a myriad of different chemical marks at hundreds of thousands of genome locations in different cells at different stages of differentiation. But what determines the site-selectivity of these chromatin remodeling complexes, how is the position of the nucleosomes regulated and what is the molecular basis of environment-epigenome interactions? The answer to all of these questions is RNA, says Mattick.
The intrinsic nucleic acid nature of ncRNAs gives them the dual ability to function as ligands for proteins and mediate base-pairing interactions that guide ncRNA containing complexes to specific RNA or DNA target sites. In addition, lncRNAs can fold into complex secondary and higher order structure to provide greater potential for protein and target recognition. Moreover their flexible and modular scaffold nature enables lncRNAs to tether protein factors that would not interact or functionally cooperate if they only depend on protein-protein interactions (see [12]).
The evidence so far indicates the most nuclear lncRNAs function by guiding chromatin modifiers to specific genome loci. Many lncRNA-mediated mechanisms of gene regulating have been identified also in the cytoplasm. These often show sequence complementarity with transcripts that originate from either the same chromosomal locus or independent loci. Upon recognizing the target by base pairing they can modulate translation either positively or negatively, or it can increase or decrease mRNA stability, thereby affecting translation.
lncRNA are much more tissues and cell specific than proteins. The emerging picture suggests that every single cell in the body may be uniquely defined by its regulatory lncRNAs, which in turn recruit transcription factors, take part in modifying chromatin structure, and modulating gene expression.
There are many classes of small RNAs, the best characterized is microRNAs (~23 nt) that regulate gene expression through RNA interference by complementary base pairing to RNA or DNA. They are transcribed from the genome as primary miRNA transcripts that are processed in the nucleus and later in the cytoplasm by the RNAse III enzymes Drosha and Dicer respectively. Typically one strand of the mature miRNA duplex then associates with the RNA-induced silencing complex (RISC) and interacts with its target [16]. More than 1 700 miRNAs have been identified ‘with high confidence’ over 38 genomes so far [17]. They regulate most mRNAs and possibly lncRNAs to influence almost every facet of animal and plant development and many aspects of brain function, and are often dysregulated in cancer and other complex diseases [9].
Related animal-specific small RNAs (24-30 nt) siRNA also involve RNA interference. piRNAs (piwi-interacting) are involved in silencing transposons mainly in the germline. Small nucleolar (sno)RNAs, which may be spread by retrotransposition, are derived from the introns of protein-coding and non-coding host transcripts; they guide specialized protein complexes to impart sequence-specific 2’-O-methylation (box D/D snoRNAs), or the isomerization of specific uredines to pseudouridines of target RNAs, and are usually localized in the nucleolus, a small dense body inside the nucleus. A related class (box H/ACA snoRNAs) is found Cajal bodies in the nucleus and use a specific localization signal also found in telomerase RNA. Originally thought to modify ribosomal RNAs for translation, some show imprinted, tissue-specific and/or context-dependent expression, especially in the brain, and also target other RNAs including small nuclear RNAs (snRNAs), tRNAs and possibly even mRNAs.
A new class of animal-specific nuclear tiny (ti) RNAs are derived just downstream from transcription initiation sites, and thought to mark the position of nucleosomes, which are preferentially positioned at exons in somatic cells and germ cells, providing a mechanism for epigenetic inheritance.
Another class of small RNAs locate to splice sites (~17-18 nt). These splRNAs are derived from the 3’ end of exons, whereas tiRNAs are posited on the 5’ side of the first nucleosome. These loci specific regulatory RNAs may be much more common.
RNA can also be modified by methylation. RNA methyltransferase plays a role in the development of the brain and other organs and is required for retrotransposon silencing in somatic cells in Drosophila. SnoRNAs, snRNAs and tRNAs are cleaved at specific positions to produce smaller RNAs that have further regulatory functions.
RNA is subject to a great deal of context-dependent editing, especially in the brain [9]. Base-deamination is catalyzed by two classes of enzymes in animals: ADARs (adenosine deaminases that act on RNA) change adenosine to inosine (A>I), which behaves like guanosine but has different base pairing qualities; and APOBEC (ApoB editing complex), which are vertebrate specific and change cytosine to uracil, and may act on RNA or DNA. There are 3 ADARs in animals: ADAR1 and ADAR2 are found in vertebrates and invertebrates, and expressed in most tissues, but particularly highly expressed in the nervous system. Loss of these genes in mice leads to embryonic or postnatal death. ADA3 is vertebrate- and brain-specific, but little is known about its function.
A>I editing occurs in thousands of transcripts. Most of the edited sites occur in non-coding regions, implying that editing not only alters proteins, but also RNA-based regulatory circuits, and hence potentially central to learning and brain function. There is a massive 30 times increase in RNA editing in humans compared to mouse; more than 90 % occurring in primate-specific Alu sequences, which evolved from a functional RNA ancestor, the 7SL RNA of the signal recognition particle. Alu sequences invaded the primate lineage in three successive waves and now comprise ~ 1.3 million mostly sequence-unique copies collectively accounting for ~10.5 % of the human genome. A>I editing also increased during primate evolution, and new editable Alu insertions after the human chimpanzee split are significantly enriched in genes related to neurological functions and neurological diseases.
The ABOBECs were discovered by their action on Apolipoprotein B mRNA, where a C>U editing introduced a stop codon resulting in a truncated protein in intestine as opposed to the longer form in liver. There are 5 families of APOBECs, two of which, APOBEC1 and 3 specific to mammals. The best characterized enzyme is AID, which is involved in somatic rearrangement and hypermutation of immunoglubulins. AID appears to edit DNA but may be targeted by RNA. AID deaminates 5’methylcytosine to form thymine and is required for the reprogramming of cells to pluripotency. APOBEC2 is required for muscle differentiation.
There are many parallels between the nervous system and the adaptive immune system, including immunoglobulin domains in many neuronal cell surface receptors, suggesting that the adaptive immune system evolved in vertebrates from the nervous system, and both may use similar mechanisms to tune receptor interactions. Moreover, numerous unusual DNA repair enzymes are present, many of which appear linked to reverse transcriptase activity. This suggests that RNA-directed DNA recoding may play a role in long-term memory formation, and possibly inheritance of acquired intelligence, as suggested by me, (see [18] Rewriting the Genetic Text in Human Brain Development, SiS 41), not Mattick.
The APOBEC3 family originated after the divergence of the marsupial and placental lineages and has greatly expanded in the primate lineage. At least some of the members as well as APOBEC 1 appear to be involved in the control of exogenous and endogenous retrotransposition, possibly by inhibiting reverse transcription. Al alternative possibility is that these enzymes (one of which APOBEC3G expressed in neurons) have evolved to domesticate transposition. De novo LI retrotransposition events occur in neural progenitor cells and may therefore contribute to individual somatic mosaicism in the brain. This process appears to be regulated by Wnt signaling pathways and transcription factors known to be important in neural differentiation. Transposon mobilization may not simply have played a role in genome evolution but also in real time genome dynamics that enable the extraordinary in situ evolution of functional complexity in the neuronal networks of the human brain.
Mattick remarked [9, p. 1610]: “most of the information [in the human genome] is involved in complex regulatory processes that underpin development and brain function. This includes the vast numbers of non-coding RNAs and transposons, which rather than being junk, appear to provide the regulatory power and plasticity required to program our ontogeny and cognition.”
Article first published 16/06/14
Comments are now closed for this article
There are 4 comments on this article.
Rory Short Comment left 17th June 2014 06:06:55
Right from my first hearing about GM, particularly because of its commercial promotion, my gut feeling was that its commercialisation was wrong and articles, such as this one, now provide the evidential backing for my gut feeling of way back then.
Todd Millions Comment left 18th June 2014 15:03:42
Facinating-almost as if life was condensed from an encyclopidia of all possibilities.
This brings to mind Holye's seeding of life from other places too the earth,the possibilities hitching along for the ride.Could these DNA/RNA interactions explain why out of africa "EVE" lineage models failured too predict Deneousnian strains in modern humans?
David Collier Comment left 21st June 2014 14:02:10
Thank you Dr Ho for this information. If this area of knowledge continues to expand the human race will no longer be able to avoid responsibility for its own psychosomatic diseases, as mentioned in the Bible and the Bhagavad Gita ('As a man thinketh in his heart so he is').
Further, this level of physiological sophistication in the human body stretches the credulity of a Darwinian evolutionary explanation with no element of intelligent design. Animals do not seem to have the comprehensive executive function enabled by the human neocortex; arguably the human body is a teaching machine in terms of the personal consequences of particular thoughts and attitudes.
Ryan N Comment left 27th June 2014 22:10:15
Rory, nothing mentioned in this article about gmo's. Fantastic summary of the latest in genetics and our understanding of the microcosm of life at the genetic level. Thank you!