Water's Quantum Jazz
Other reports in this series
Water has a special dynamic relationship with ions dissolved in it that will determine their chemistry in the cell Dr. Mae-Wan Ho
The interaction of water with ions has been studied for over a century, but much remains to be understood. In general, ions are classified into two groups - ‘kosmostopes’ or ‘chaotropes’ - according as to whether they induce order or disorder in water [1].
Thus, Li+ (lithium) dissolving in water results in a large negative entropy change, as typical of a kosmotrope, while Cs+ (caesium) dissolving is accompanied by a positive entropy change, as appropriate for a chaotrope. Another measure is viscosity, which increases with lithium and decreases with caesium.
Both types of ions are generally surrounded by water molecules forming a ‘solvation shell’ that shield their charges from other ions. The strength with which the ions bind to their solvation shell is expressed in terms of activation energy, the energy needed to strip a water molecule away from the ion relative to that needed to strip a water molecule from another water molecule. Kosmotropes are strongly bound to their neighbouring water, and therefore have large positive activation energies, as distinct from chaotropes that are more weakly bound.
In general, the solvation shell consists of a single layer of water molecules hydrogen-bonded to the ion. A strong kosmotrope could have up to 5 or 6 water molecules in its solvation shell. A strong chaotrope, on the other hand, could bind a single water molecule, or none at all, as it approaches the limit of a nonpolar solute, where the surrounding water molecules only form hydrogen bonds with one another, enclosing the solute in a cage-like clathrate structure (see [2] Cooperative and Coherent Water, SiS 48).
How rigid is the solvation shell around the solute, and does the hydrogen bond - which breaks and reforms within a picosecond (10-12s) - readily change partners or orientate?
Molecular dynamic simulations carried out on NaCl solutions by researchers at Ecole Normale Superieure Paris, France, and University of Colorado, Boulder, Colorado, USA, showed that the hydration sphere around chloride ions is labile, consistent with previous assessment of chloride as a weak chaotrope [3]. A molecule in the first solvation shell can switch partners by a large reorientation - an ‘angular jump’ of some 60 to 70˚ - after which the water molecule involved leaves the anion’s solvation shell and moves to the second shell (see Fig. 1).
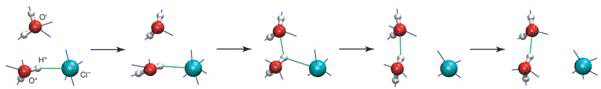
Figure 1 A water molecule switching partners in hydrogen-bonding between the ion and another water molecule
This will, no doubt, contribute to the challenging study of water dynamics in the hydration shells of biomolecules such as proteins and nucleic acids, where both experiments and simulations have already pointed to a dynamical regime quite different from bulk water. In particular, it suggests that the first hydration shell is very labile, exchanging rapidly with the second shell and not rigid as has been previously suggested
Researchers led by KJ Gaffney at Stanford University California in the USA and Stockholm University in Sweden decided to put the simulation result to experimental test. They devised a variant of two-dimensional infrared spectroscopy to investigate this jump in orientation that occurs on switching partners in hydrogen bonding. The results confirmed that a water molecule can shift its donated hydrogen bond between water and perchlorate acceptors by large angular jumps of 49 + 4˚ on average.
Actually, changing partners involves the water molecule doing a swivel dance in two steps: a really fast swivel of 40˚ in about 50 fs (femtosecond, 10-15s), followed by a slower rotation of 27˚ in 1 about ps.
Dissolving sodium chlorate NaClO4 in water mixed with heavy isotope deuterium tracer (D2O) makes it possible to distinguish two kinds of OD bonds by the way they stretch when excited by infrared light. The OD group acting as donor to another water molecule (ODw) absorbs at 2 534 cm-1, whereas the OD group donating to a perchlorate anion (ODp) absorbs at 2 633 cm-1. Thus, one can track hydrogen-bond exchange in two-dimensional infrared spectroscopy. In addition, the orientation jump angle can be measured using polarized probes that make the signal from the exchanged hydrogen bond highly dependent on the polarization direction. That was how the researchers managed to catch this exotic dance step in water’s quantum jazz
What happens when more than one kind of ions exist in solution as is usually the case in real life?
Researchers at the Institute for Atomic and Molecular Physics in Amsterdam, Netherlands, used a combination of Terahertz dielectric spectroscopy and femtosecond infrared (fs-IR) spectroscopy to study water dynamics around different ions: magnesium, lithium, sodium and caesium cations, as well as sulphate, chloride, iodide and perchlorate anions. They found that the effect of ions and counterions on water can be strongly interdependent and nonadditive, and in certain cases, extends well beyond the first solvation shell, the layer of water immediately surrounding the ion.
Terahertz dielectric spectroscopy involves characterizing the propagation of a Terahertz (1012 Hz) pulse lasting ~1ps through the salt solution. The pulses are delayed by refraction (bending of electromagnetic waves) and diminished by absorption. In water, a marked frequency dependence of refraction and absorption arises when the dipoles of water molecules fail to keep up reorienting with the externally applied oscillating field. This leads to a large absorption peak at GHz (109 Hz) and a smaller one around 0.6 THz for pure water at room temperature. These peaks are attributed respectively to the collective reorientation of water molecules (relaxation time tD ~8ps), and the reorientation of partly hydrogen-bonded individual water molecules (relaxation time t2 ~250 fs). As the result of interaction with solvated ions, the reorientation of water molecules around ions slows down, shifting the absorption peak to lower frequencies and reducing the absorption at THz frequencies. This is referred to as depolarization.
. The hydration number Np is the number of moles of slow water dipole per mole of dissolved salt and is directly proportional to the slopes of the lines plotting depolarization against concentration (Fig 2 left). Generally, ions with a high charge density (small, multivalent ions with more than one charge) affect the dynamics of a large number of water molecules (high hydration number); these correspond to the kosmotropes described earlier. Ions with a lower charge density (large monovalent ions with only a single charge) tend to affect a small number of water molecules and have a small hydrations number; these correspond to the chaotropes.
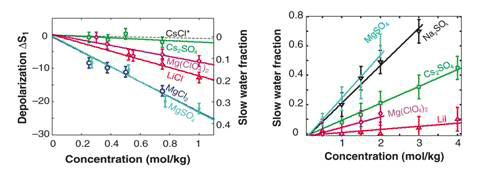
Figure 2 Dielectric relaxation (left) vs femtosecond infrared spectroscopy (right)
The results for MgCl2, LiCl and CsCl are in good agreement with this general rule. For Mg(ClO4)2, Np = 6 because ClO4 is a very weakly hydrated anion, the observed slowly reorientating water molecules are likely located in the first solvation shell of the Mg2+ ion. In contrast, for Cs2SO4, Np is only 1. Apparently, the water molecules in the solvation shell of Cs+ show reorienting dynamics similar to those of the bulk water, probably because the positive charge of the Cs+ ion is distributed over a large volume. The slowly reorienting water can be attributed to a water molecule located within the solvation shell of strongly hydrated SO42+ ion, probably forming hydrogen bonds, with both OH groups, to two oxygen atoms of the SO42+ ion. The low values of Np found for Cs2SO4 indicates that the effect of anions on water reorientation is either negligible or not measurable by dielectric spectroscopy.
Fs-IR spectroscopy probes the reorientation dynamics of individual water molecules with high time resolution. In these experiments, water is doped with 4 percent of D2O. The OD-stretch vibration is excited, and molecules with their OD group preferentially aligned along the polarization axis of the excitation pulse are most efficiently tagged. By using a second laser probe pulse to interrogate the number of tagged molecules parallel and perpendicular to the excitation axis, the rotation of tagged molecules can be tracked.
Remarkably, MgSO4 shows a very slow reorientation component, whereas Mg2+ and SO42- individually, in combination with other ions do not. This demonstrates that the effect of ions on water dynamics can be non-additive, which needs to be explained.
Also in need of an explanation is an apparent discrepancy between the dielectric measurements and the fs-IR measurements. For hydrated cations, the two methods give different results: Mg2+ and Li+ show large immobilized fractions when measured with dielectric spectroscopy (Fig. 1, left), but the fs-IR measurements of Mg(ClO4)2 and LiI show almost complete reorientation, with a negligible slow fraction (see Fig. 1, right).
The differences between the results of dielectric relaxation and fs-IR spectroscopies can be understood by noting the different vectors that the two techniques probe: the permanent dipole moment of water p in the case of dielectric relaxation and the OD-stretch transition dipole moment m in the case of fs-IR spectroscopy (Fig. 3).
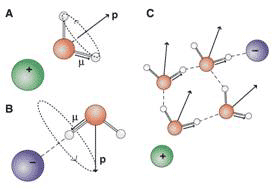
Figure 3 Cooperative dynamics of water induced by ions
The local electric field around the ions causes the dipole vector p of water molecules in the solvation shell of a cation to point away from the cation (Fig. 3A), whereas for an anion, one of the OH groups of a hydrogen-bonded water molecule points linearly towards the anion (Fig. 3B). For cations, the rotational motion of water molecules that changes the dipole vector m in the solvation does not lead to reorientation of the vector p. In the case of anions, the reverse effect occurs. the motion of p is unrestricted within a cone with fixed axis m, This explains the insensitivity of dielectric spectroscopy towards anionic hydration, and fs-IR spectroscopy towards cationic hydration. For both cations and anions, these observations lead to a molecular picture of semi-rigid hydration, i.e., water molecules in ionic solvation shells that reorient in a propeller-like manner, giving rise to partial reorientation along a distinct axis. This picture holds for salts for which one of the counterion is weakly hydrated. However, when both ion and counterion are strongly hydrated, the effect on water dynamics can be much stronger and nonadditive.
The dielectric spectroscopy data show that Mg(ClO4)2 has a hydration number Np=6 and that Cs2SO4 has a hydration number Np=1; these values are associated with water molecules directly adjacent to the Mg2+ ion and a water molecule hydrating the SO42- ion respectively. For MgSO4, Np=18, much larger than the sum of the hydration numbers of Mg(ClO4)2 and Cs2SO4; and this is because of a cooperative effect between the cation and the anion. The size of most ions allows them to be structurally surrounded by four to six water molecules. Hence a value of Np>6 and Nm>12 implies that the effect of the ion on the orientational dynamics of water extends well beyond the first hydration shell of water molecules.
The same cooperativity is observed in the fs-IR measurements where MgSO4 has a much larger fraction of slowly reorienting water molecules, corresponding to a hydration number Nm = 32; whereas Mg(ClO4)2 and Cs2SO4 have hydration numbers Nm of 4 and 9 respectively. For MgSO4, there are about twice as many slowly rotating OH groups as slowly rotating dipoles, which indicates that the same collection of slow water molecules is observed by fsIR and THz DR spectroscopies. Even the combination of moderately strongly hydrated cation Na+ with the strong anion SO42- is observed to affect the dynamics of a large number of water molecules.
The cooperativity in ion hydration can be explained by the fact that the cation and anion lock different degrees of freedom of the water molecules. The nearby presence of both ions can thus lead to a locking in both directions of the hydrogen-bond structure of several intervening water layers, giving rise to slowed water molecules well beyond the first salvation shell (see Fig.2 C). The solvation structure is expected to be quite directional between the ions. This interpretation also means that the slowly reorienting water molecules are not arranged in a spherically symmetric way around the ions.
These findings have large implications on water dynamics within the living cell that contains a huge variety of molecular and ionic species all participating in water’s quantum jazz. We can get an inkling of that from the effect of inorganic ions on proteins in solution [5] (The Rainbow Ensemble, SiS 48).
Article first published 28/09/10
Comments are now closed for this article
There are 2 comments on this article.
Michael Leonard Comment left 2nd October 2010 21:09:45
Dear all at I-S-I-S,
Thank you for the years of work you have put into discovery and release of all of this most valuable material.
I have not, as yet, been able to afford membership to help with your costs, but would like to help in other ways.
Sometimes there are some typos and formatting errors. These can slip past the tired proof-reader; especially when the material is novel, complex and full of new terms to most of your readership.
For your readership these typos interrupt the sorting of this data, and unless the reader is very determined, may become the cause of abandoning the effort.
Under is a list of those found in this article (Dancing with Ions).
Square brackets surround the possible typo.
followed by a slower rotation of 27˚ in [1 about] ps
What happens when more than one kind of ion[s] exist[ ] in solution as is
[.] The hydration number Np is the number of moles of slow water dipole[] per
m in the case of fs-IR spectroscopy (Fig. 3) [Font used for "m" is not the same as that used for "p". The text had to be zoomed to be legible.]
picture holds for salts for which one of the counterion[] is weakly hydrated.
This whole series on the wonderful attributes of water brings fresh material with which to reconsider the first chapter of Genesis.
patrons99 Comment left 5th October 2010 16:04:59
Well said, Michael - "This whole series on the wonderful attributes of water brings fresh material with which to reconsider the first chapter of Genesis." I completely agree. What exactly did God create in Genesis...the water molecule 'plus'? It's still a very BIG leap for life to arise in a primordial soup. It should give the Neo-Darwinists a more formidable task, too. For me, it's impossible not to invoke a greater power, i.e., the hand of God. The effect of inorganic ions on proteins in solution cannot be underestimated. The zeta potential of our flowing blood is important in assuring its colloidal stability, to prevent red blood cells from sludging, gel formation, or clotting. In a non-flowing situation, e.g. intracellularly, the effect of inorganic ions on proteins would have to be substantial, too. Consider the in vivo effects of Aluminum (3+) salts, Mercury (2+) salts, and Fluoride (1-) salts, all three of which are biotoxic, by mechanisms which are not fully understood. I seem to recall that fluorine can act as a hydrogen bond acceptor. I'll bet that all three of these ions interfere with water's quantum jazz, in a way that God never intended.