Water's Quantum Jazz
Other reports in this series
Macromolecules need lots of water to function effortlessly and acquire completely new talents when fully hydrated Dr. Mae-Wan Ho
Organisms have an enormous repertoire of chemical reactions that enables them to transform energy and materials for growth, development, and to do all that’s required of being alive. Perhaps most remarkably, these chemical reactions are catalyzed by specific enzyme proteins that accelerate the reaction rates by a factor of 1010 – 1023. But the question of how enzymes work remains unanswered to this day [1].
One lead player that has received far too little attention until relatively recently is water. It is well known that enzymes and other macromolecules, DNA and RNA, need a minimum amount of water in order to work at all, and much more to work efficiently. That is why cells are loaded with water, some 70 percent by weight. In terms of number of molecules, water far outnumbers all other chemical species - ions, small organic molecules and macromolecules – added together.
Water is needed for macromolecules to become flexible, so they can dance freely to water’s quantum jazz, in order to accomplish their otherwise impossible tasks of making sluggish chemical reactions happen spontaneously and effortlessly.
Many scientists are indeed coming around to the view that proteins act quantum mechanically, or, as some of us have proposed years ago, enzyme proteins are quantum molecular machines that transform energy coherently (see [2] The Rainbow and the Worm, The Physics of Organisms, I-SIS publication). But we must backtrack to sketch the whole picture.
Within the past 20 years, water has gained recognition as an active constituent of cell biochemistry and not just an inert solvent [3]. Water has a special relationship with proteins in the protein’s ‘hydration shell’. The hydration shell can be defined as water associated with the protein at the hydration end point, when further addition of water produces no change of its essential properties; in the case of an enzyme, this would be its enzyme activity. This hydration shell is a single layer of water molecules covering the protein surface. Water outside the monolayer is perturbed to a significantly smaller extent, typically not detected by measurements of properties such as heat capacity, volume or heat content (though it is increasingly recognized by newer measurement techniques, see later). The hydration shell is about 0.2g/g dry protein. The activity of lysozyme, for example, closely parallels the development of surface motion as hydration increases, which is thus responsible for the function of the protein.
In contrast to bulk water, protein-hydration water does not freeze at 0 ˚C, but can be supercooled right down to a glass transition [4] at Tg ~ 170 K (≡ -103 ˚C), and is reflected in discontinuities in the specific heat and thermal expansion coefficient of the hydration water. Below that temperature, the hydration water freezes into a glass, a non-crystalline solid that does not have a defined molecular order. Near Tg, the movement of water is arrested.
The protein however, has its own dynamic transition, an abrupt onset of atomic motions that can be detected by neutron scattering, and occurs at TD ~225 + 5 K. This is a generic property of hydrated proteins, and is absent in dehydrated systems. It is therefore related to the dynamics of the hydration shell. Some researchers regard the protein dynamic transition as the “microscopic manifestation” of the glass transition of the hydration shell. Others, however, interpret it as a liquid to liquid transition, from a fluid, high density liquid (HDL) to the less fluid, low density liquid (LDL), or supercooled water [3].
Similar behaviour was found for confined water in various biological and non-biological environments, and a range of 18 solutions including low molecular weight organic glass formers, polymers, sugars as well as protein and DNA probed with broad band dielectric spectroscopy (see Box) between 10-2 and 107 Hz [5]. Universal features in the dynamics of water appeared both above and below the glass transition, which were similar to supercooled water and supercooled confined water respectively. However, the glass transition temperature spans a rather broad range from 165 to 220 K.
Dielectric spectroscopy
A dielectric is a molecule such as water that is polarized - with separated positive and negative charges - or can be polarized by an applied electric field. Dielectric spectroscopy measures the dielectric response of a sample to an external electromagnetic field applied over a range of frequencies. The dielectric property of a material is expressed in a complex number known as permittivity. The real part of permittivity measures the energy stored, the imaginary part measures the energy loss and is called the dielectric loss. When the external field is applied, dipoles in the sample will orient with the applied field. At low frequencies, they can follow the polarization of the applied field perfectly, resulting in maximum permittivity and minimum dielectric loss. As the frequency increases, the molecules can no longer keep up with the changing electric field, this results in less energy stored and higher losses. At even higher frequencies, the molecules no longer respond to the applied field, and the molecules come apart. For example, water molecules are ‘stretched’ at frequencies above 1011 Hz, and beyond that, they break apart.
Dielectric relaxation time is a measure of the time it takes for separated charge in a dielectric to become neutralised by conduction process.
Within the past several years, a new Terahertz (1012 Hz) spectroscopy has created a stir in the protein hydration community. Martina Havenith and colleagues at Ruhr University Bochum, Germany, have been using this new ‘table-top’ technology to reveal global properties of proteins and their hydration shells [6], prompting a number of water scientists to rethink protein hydration.
Many sophisticated techniques have been used to probe the dynamics of proteins and hydration water over a wide range of time and space. For example, NMR relaxation spectroscopy resolves water dynamics from nanoseconds to seconds whereas X-ray crystallography reveals fixed protein structures and bound water molecules in the protein interior and vicinity. Both techniques provide information on the short range hydration up to 3 Å (angstrom, 10-10 m) from the protein surface which corresponds to one hydration layer. Traditional dielectric spectroscopy probes dynamics from 100 seconds down to 100 ps (picoseconds, 10-12 s). Terahetz dielectric spectroscopy extends the time scale down to ps range. Neutron scattering resolves ps dynamics. On the fastest time scales, two-dimensional infrared spectroscopy probes processes that take place in femtoseconds (1 fs =10-15 s). And the remarkable picture emerging is that protein and hydration dynamics may be correlated over all time scales, as indeed they would be in quantum coherent systems [2].
Terahertz absorption spectroscopy, made possible by powerful ‘table-top’ Terahertz electromagnetic sources, opens up a new window between microwaves and infrared to peer into the global interactions of water with proteins that are fully dissolved (in excess water).
The absorption of the solvated protein increases linearly with frequency in a narrow frequency range of 2.25-2.55 THz. Concentrating on measurements at a single frequency, the researchers found that the absorption coefficient of the solution relative to bulk water increases with protein concentration before dropping and decreasing almost linearly at higher concentrations. The concentration dependence of the THz absorption gives similar curves at three different temperatures 15, 20 and 22 ˚C (see Fig.1, left).
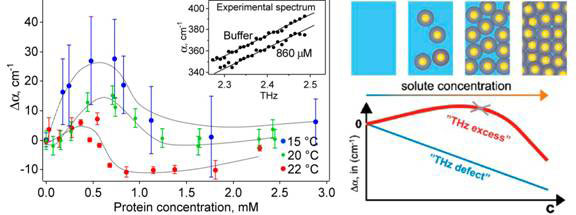
Figure 1 Absorption coefficient vs protein concentration (left) and it explanation (right)
At 0.5 to 1 mM protein concentration, when the absorption coefficient peaks and begins to fall again, the volume of water displaced by the protein molecules is negligible at around 1 percent. Consequently, the hydration water around the protein must be contributing substantially to the total THz absorption.
The researchers resorted to molecular dynamic simulations to compute the absorbance of the protein and the first hydration layer as a function of the distance between protein surfaces, which would depend on the protein concentration. In agreement with experiment observations at concentrations beyond the peak of absorbance, the distance between the proteins significantly influences the absorbance of the protein and its first hydration layer and layers beyond. The absorbance decreases as the proteins are brought closer together from 24 to 18 A, then flattens for the shortest distances, changing little with distance between proteins.
Further molecular dynamic simulations revealed that the hydrogen bonds between water molecules in hydration layers up to about 10 Å survive longer than those in bulk water. The experimental data indicate an average separation of >20 Å when absorbance peaked, spanning some 7 layers of water.
The initial findings were made in a genetically engineered protein l*6–85, but similar results were obtained with natural lysozyme and myoglobin [7]. At 23 g water per g protein of lysozyme, absorbance surged upwards with increased structural flexibility of the protein. Myoglobin, similarly, exhibited a maximum at 98 percent by weight of water (24g/g).
While bulk water and its absorption decrease with increasing protein concentration, this is more than offset by the absorption of hydration layers and the hydrated protein, which becomes a dynamically coherent unit (Fig. 1, diagram on the right).
The unexpected findings in Terahertz dielectric absorption spectroscopy [6, 7] have stimulated researchers to examine the electrostatic properties of the hydration water. David leBard and Dmetry Matyushov at Arizona State University, Tempe, USA, said that [8] to explain those observations requires a very large “effective dipole moment of the protein and its hydration shell, much exceeding the dipole moment of the protein itself.”
LeBard and Matyushov showed by means of numerical simulations that protein hydration waters are polarized into a ferroelectric shell some 3-5 water molecules thick with large average dipole moment. Moreover, the dipole moment of the hydration shell fluctuates with large amplitude, much bigger than those in bulk water, and far exceeding the magnitude prescribed by the usual linear response theory, as also demonstrated in real measurements. These large fluctuations are dominated by slow nanosecond dynamics probably associated with collective conformational movements of the protein.
These findings are just what is expected from the dynamic liquid crystalline nature of living organisms discovered in my laboratory in 1992 implying that water associated with macromolecules are also polarized and moving coherently with the macromolecules [2].
Is there any evidence that the protein-hydration system is quantum coherent, as I have suggested the organism is quantum coherent?
The possibility of quantum tunnelling has been invoked in enzyme catalysis. Quantum tunnelling is a quantum mechanical effect in which particles go under an energy barrier to achieve a chemical reaction that’s impossible in classical chemistry. Quantum tunnelling is already well accepted in electron transfer and frequently observed for proton transfer [2].
Recent computational and experimental studies have clearly shown that the flexibility of the proteins induced by water is important in reducing free energy barrier between reactants and products. The flexing of proteins increases the probability of quantum tunnelling between reactants and products by a transient compression of the energy barrier. (Obviously, in the case of proton or electron transfer, quantum tunnelling would only take place if the actions of donor and acceptor are also correlated or coherent, so coherence must extend to the ensemble of donor and acceptor proteins.) The same studies indicate that the dynamics of proteins are closely tied to that of water. Some scientists would go as far as to say it is mainly the mobility of water that determines the magnitude of protein fluctuations, not only at the protein surface, but also in the protein core. But then the collective, conformational movements of proteins would feedback to influence the hydration water coherently (see above).
One sign of quantum effect is the difference in behaviour observed when hydrogen (H) is substituted with its heavy isotope deuterium (D), as already clearly indicated for water itself (see [9] Cooperative and Coherent Water, SiS 48).
Fabio Bruni and colleagues at University Studi Rome Tre in Italy used dielectric spectroscopy to compare the dielectric relaxation of lysozyme in ordinary water (H2O) and in heavy water (D2O) in the frequency range of 10-2 to 107 Hz, and over the temperature range 210 K to 330 K [10].
If the effect is classical, there should be no difference between the sample in H2O or D2O. However, large differences were found. In particular, the dielectric relaxation times of H2O were completely different from those measured with D2O over the entire temperature range (see Fig. 2).
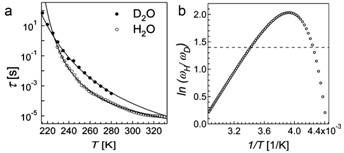
Figure 2 Dielectric relaxation time t vs temperature T (a); natural logarithm of the reciprocal of relaxation times ratiotD/tH (≡wH/wD) vs reciprocal of temperature; the horizontal dotted line marks the value 1.4, which should hold for all temperatures if there were no quantum effects
These observations were in close agreement with results of a sophisticated theoretical study using quantum mechanical methods from first principles.
In addition, the researchers reanalysed data from a deep inelastic neutron scattering experiment, which measured momentum distribution of protons n(p) and mean kinetic energy for a lysozyme sample prepared the same way as for the dielectric spectroscopy experiment. Measurements were made both above and below the protein’s dynamic transition temperature (see above) at 290 K and 180 K respectively. They computed the minimum fraction of protons required to produce the observed difference in the distribution of momentum between the two temperatures, assuming that the mean kinetic energies of the proton is independent of temperature except for the small kT (Boltzmann, background) contribution; and that the momentum distribution at 290 K arises from a population of protons with the same n(p) as that at 180 K plus a contribution due to an unknown fraction of protons showing a characteristic quantum mechanical behaviour. This fraction of protons that showed quantum behaviour turns out to be 0.29, precisely the fraction of protons present in the sample that belong to the hydration water.
The fascinating story of DNA and water is just beginning. Hydrating shells of DNA share many of the properties of hydration shells of proteins. In addition, water turns DNA into a electrical conductor and gives it magnetic properties; and many are looking into exploiting synthetic DNA in new molecular electronic devices.
Hydration water of DNA appears to have quite unusual dynamics as measured by time-resolved Stokes-shift (TRSS) and confirmed by molecular dynamics simulation. In the TRSS experiments on a 17 base pair oligonucleotide, light excites the DNA and is re-emitted as fluorescence, shifted to lower energy compared to the light absorbed (Stokes shift). The mean fluorescence frequency is measured as a function of time after excitation, covering six decades from 40 fs to 40 ns.
Most unexpectedly, the relaxation (decay of fluorescence) did not show distinctive time scales that could be easily attributed to multiple, independent processes. Rather it fits a single power law, suggesting a high degree of correlation over all time scales.
Researchers led by Mark Berg at South Carolina University in the United States used a polarization model based on the idea that the coupling between components in the system is due to polarization of one component by another, which not only fit the experimental data well, but also enabled them to assign the TRSS response to different components of the system: the water, the counter-ions of DNA or the DNA molecule itself [11]. The results showed that water is the dominant contributor to the TRSS response at all times. Its relaxation spans the entire measured time range and accounts for the power law behaviour. Counter-ions have a secondary, but non-negligible contribution with a well-defined relaxation time of about 200 ps. The DNA relaxation time is near 30 ps, but the amplitude is very small.
The ability of DNA to conduct electricity has remained controversial for years. But Christophe Yamahata and colleagues at Tokyo University and Kagawa University Takamatsu in Japan have convincingly demonstrated that the ability of DNA to transport electrical charge depends on water [12]. DNA bundles suspended in air between nano-tweezers (see Fig. 3) increased exponentially in electrical conductivity up to 106 fold as the relative humidity of the air increased. The increase was attributed to an increase in the concentration of charge carriers as DNA hydration rose with relative humidity; the charge carriers being H+ and OH-, suggesting that proton (positive electricity) jump conduction could be involved (see [13] Positive Electricity Zaps Through Water Chains, SiS 28) as well as electron conduction.

Figure 3 DNA suspended in air with nano-tweezers
An important finding is that the pathway for charge transfer (conduction) is through the stacked bases in the core of the double helix where the p molecular orbitals overlap. A molecular orbital is a waveform describing the distribution and energy of a pair of electrons, and is most commonly represented as a linear combination of atomic orbitals of atoms forming a covalent bond in the molecule. A p orbital is the molecular orbital of a p bond where two lobes of the atomic orbitals overlap and join up. The halves of a molecule joined by a p bond cannot rotate about that bond without breaking it. Carbon and nitrogen compounds form p bonds when they engage in multiple bonding, as in C=C and N=N. In ring compounds such as benzene and bases in DNA, where double and single bonds alternate, the p bond electrons are delocalized, or spread over the entire ring structure and stabilizes it.
Julia Berashevich and Tapash Chakraborty at the University of Manitoba, Winnipeg, Canada, used quantum chemical calculations to demonstrate that interaction of the bases with water (see Fig. 4) breaks some of the p bonds in the ring structure of the bases, giving rise to unbound p electrons. These unbound electrons, together with the change in DNA conformation from A (dehydrated) form to B (hydrated) form, could account for the exponential increase in conductivity as relative humidity and hydration of the DNA molecule increases.
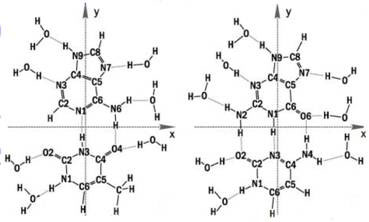
Figure 4 Water binding to DNA bases
Furthermore, at low temperatures, the efficiency of charge transfer is determined by the spin interaction of two unbound electrons located on neighbouring bases of the same strand. Exchange is allowed only when the electron spins are antiparallel (in opposite directions). Hence the conductance of DNA can be controlled by a magnetic field. This gives the potential for developing nanoscale ‘spintronic’ devices based on the DNA molecule, where the efficiency of spin interaction will be determined by the DNA sequence.
Regardless of the practical applications, these findings have profound implications for the biological functions of DNA, apart from serving as a linear code for the sequence of amino acids in proteins. We are only touching the tip of a very large iceberg.
Article first published 18/10/10
Comments are now closed for this article